FDA BRIEF: Week of June 13, 2016

ASPIREASSIST
Aspire Bariatrics, King of Prussia, PA, USA

INDICATION FOR USE: Intended to assist in weight reduction of obese patients. It is indicated for use in adults aged 22 or older with a Body Mass Index (BMI) of 35.0-55.0 kg/m2 who have failed to achieve and maintain weight loss with non-surgical weight loss therapy. Intended for a long-term duration of use in conjunction with lifestyle therapy and continuous medical monitoring
UNMET NEED: Need for approach for effective control of calorie absorption
REG PATHWAY: PMA, Device Procode: OYF
DEVICE DESCRIPTION:
Designed to facilitate weight loss in obese patients by
- enabling removal of a portion of stomach contents through a gastrostomy tube
- requiring thorough chewing in order for stomach contents to enter the 6mm diameter tube
Device consists of
- ‘A-Tube’ and “Gravity” flow director system through which patients aspirate (drain) gastric contents about 20 to 30 minutes after consumption of a meal directly into toilet
- Key components: A-tube, Skin Port, Skin Port Installation Toolkit, Connector, Companion, Tubing Set, Reservoir, Lanyard, Emergency Clamp
- Used after the three (3) major meals each day
- Takes 5-10 minutes to complete
- Removes about 30% of the calories consumed
- Aids in portion control; all food must be thoroughly chewed to fit through the tube
EFFECTIVENESS
- PATHWAY Pivotal Trial, prospective, randomized, multi-center, controlled, open-label, 52-week , n=111 vs 60 (control)
- Co-primary Endpoints:
- 52-week comparison of mean percent excess weight loss (%EWL), AspireaAssist (AT) vs Control Group
- AT responder rate dichotomized at 25% EWL
- %EWL: 31.5% vs 9.8% (p=0.0083)
- 25% EWL: No met
SAFETY:
- Contraindication: Uncontrolled hypertension, diagnosed bulimia, diagnosed binge eating disorder, night eating syndrome, pregnancy or lactation, inflammatory bowel disease. Also previous history of serious pulmonary or cardiovascular disease, coagulation disorders, chronic abdominal pain
- Surgical placement risks
- Risks related to the abdominal opening for the port valve
- Side effects: Occasional indigestion, nausea, vomiting, constipation and diarrhea.
VENTANA PD-L1 (SP142) Assay
Ventana Medical Systems (Roche), Tucson, AZ, USA
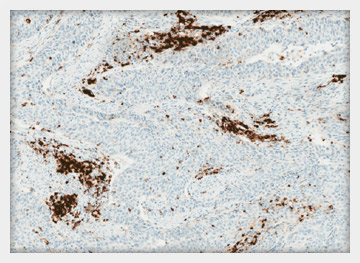
INDICATION FOR USE:
Qualitative immunohistochemical assay using rabbit monoclonal anti-PD-L1 clone SP142 intended for use in the assessment of the PDL1 protein in formalin-fixed, paraffin-embedded (FFPE) urothelial carcinoma tissue stained with OptiView DAB IHC Detection Kit and OptiView Amplifcation Kit on a VENTANA BenchMark ULTRA instrument.
PD-L1 status is determined by the proportion of tumor area occupied by PD-L1 expressing tumor-infiltrating immune cells (% IC) of any intensity. PD-L1 expression in ≥ 5% IC determined by VENTANA PD-L1 (SP142) Assay in urothelial carcinoma tissue is associated with increased objective response rate (ORR) in a non-randomized study of TECENTRIQ™ (atezolizumab).
REG PATHWAY: PMA, Device Procode: PLS
DEVICE DESCRIPTION:
Device Kit Components
- Optimized reagents to complete immunohistochemical staining procedure
- Recombinant rabbit monoclonal antibody produced as purified cell culture supernatant and contains sufficient reagent for 50 tests
- Antibody and detection reagents are provided as ready-to-use dispensers
Device Instrumentation and Software
- Assay is performed on the BenchMark ULTRA automated staining instrument using the VSS software version 12.2
- Assay protocol is assay specific
- Software designed to recognize and group the VENTANA PD-L1 (SP142) Assay, requiring that all system reagents are used together
EFFECTIVENESS:
- Non-randomized, a multicenter, open-label, trial designed to evaluate the efficacy of TECENTRIQ in patients with locally advanced or metastatic urothelial carcinoma
- PD-L1 expression in ≥ 5% IC determined by VENTANA PD-L1 (SP142) Assay in urothelial carcinoma tissue associated with increased objective response rate (ORR) from TECENTRIQ
- Performance of Assay also supported by analytical validation studies
SAFETY:
- Risks based on data collected in clinical study
- Failure of device may lead to failure to correctly interpret test result
- Testing on tumor specimens does not present additional significant safety concerns, as these samples are routinely removed for diagnosis
CEREVE Sleep System
Cereve, Pittsburgh, PA, USA

INDICATION FOR USE: Reduce sleep latency to Stage 1 and Stage 2 sleep in patients with primary insomnia
REG PATHWAY: De Novo, Class II, Product Code: PLU
DEVICE GENERIC DESCRIPTION: Thermal System for Insomnia: Prescription device for use in patients with insomnia that is used to apply a specified temperature to the skin surface
WALL FLEX Biliary Fully Covered Stent System
Boston Scientific Corporation, Marlborough, MA, USA

INDICATION FOR USE: Indicated for indwell up to 12 months in the treatment of benign biliary strictures secondary to chronic pancreatitis
REG PATHWAY: De Novo, Class II, Product Code: PNB
DEVICE GENERIC DESCRIPTION: Metallic Biliary Stent System for Benign Strictures: Prescription device intended for the treatment of benign biliary strictures. The biliary stents are intended to be left indwelling for a limited amount of time and subsequently removed. The device consists of a metallic stent and a delivery system intended to place the stent in the bile duct. This device type is not intended for use in the vasculature.