Authorizations: ORAQUICK Ebola test, TRIKAFTA for cystic fibrosis, REYVOW for migraine, SCENESSE for erythropoietic protoporphyria

ORAQUICK EBOLA RAPID ANTIGEN TEST
OraSure Technologies, Inc
INDICATION FOR USE: In vitro diagnostic single-use immunoassay for the qualitative detection of antigens from viruses within the Ebolavirus genus but does not differentiate
between these viruses
ADDRESSING UNMET NEED: First rapid diagnostic test for the Ebola Virus Disease (EVD). The test provides a rapid, presumptive diagnosis that must be confirmed.
TYPE OF DEVICE: Device to detect antigens of biothreat microbial agents in human clinical specimens.
To detect antigens of biothreat microbial agents in human clinical specimens is identified as an in vitro diagnostic device intended for the detection of antigens of microbial agents in specimens collected from patients with signs and symptoms of infection with biothreat microbial agents and who have been exposed to these agents or are suspected or at risk of exposure. The device can include antibodies for immobilization and detection of the analyte. This device may also be used for cadaver testing to prevent human diseases for which cadavers constitute a source of human-to-human transmission.
EFFECTIVENESS AND SAFETY:
- Data from multiple clinical studies of blood samples and cadaveric oral fluid from the 2014 West African outbreak and analytical studies
- Reasonable assurance of the safety and effectiveness when intended to identify antigens associated with Ebola virus in blood from symptomatic patients and oral fluid of cadavers – with high levels of Ebola virus
- OraQuick Ebola Test is not intended to be used for general Ebola infection screening (e.g., airport screening) or testing of individuals at risk of exposure without observable signs of infection
REG PATHWAY: De Novo classification
- Emergency Use Authorization (EUA) pathway -Collaboration between FDA, WHO and Democratic Republic of Congo (DRC)
- Breakthrough Device designation
- Regulation Number: 21 CFR 866.4002
- Regulation Name: Device to detect antigens of biothreat microbial agents in human clinical specimens
- Regulatory Class: Class II
- Product Code: QID

TRIKAFTA (elexacaftor/ivacaftor/tezacaftor) tablet
Vertex
INDICATION: Treatment of cystic fibrosis (CF) in patients aged 12 years and older who have at least one F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene.
If the patient’s genotype is unknown, an FDA-cleared CF mutation test should be used to confirm the presence of at least one F508del mutation.
ADDRESSING UNMET NEED: First triple combination therapy available to treat patients with the most common cystic fibrosis mutation.
MECHANISM OF ACTION: Elexacaftor and tezacaftor bind to different sites on the CFTR protein to increase the amount of CFTR protein delivered to the cell surface. Ivacaftor potentiates the channel open probability (or gating) of the CFTR protein at the cell surface. The combined effect of elexacaftor, tezacaftor and ivacaftor is increased quantity and function of F508del-CFTR at the cell surface, resulting in increased CFTR activity
EFFICACY:
- 2 randomized, double-blind, active and placebo-controlled trials, patients with CF, 12 years and older
- First trial – n=403 with F508del mutation and mutation on second allele that is not responsive to ivacaftor or tezacaftor/ivacaftor alone
- Second trial- n=107 patients with two identical F508del mutations
- Primary analysis: Increases in % predicted forced expiratory volume in one second, (ppFEV1). Trikafta increased the ppFEV1 in both trials ( 13.8% from baseline vs. placebo and 10% from baseline vs. tezacaftor/ivacaftor)
SAFETY:
- Serious adverse drug reactions: Rash and influenza (flu) events
- Most common adverse drug reactions: Headaches, upper respiratory tract infections, abdominal pains, diarrhea, rashes, increased liver enzymes (alanine aminotransferase and aspartate aminotransferase), nasal congestion, increased blood creatine phosphokinase, rhinorrhea, rhinitis, influenza, sinusitis and increased blood bilirubin
- Warnings related to elevated liver function tests
REG PATHWAY: NDA
- Priority Review, Fast Track, Breakthrough Therapy, and orphan drug designation
- Rare pediatric disease priority review voucher
- Postmarketing requirements: Rat carcinogenicity studies, hepatic impairment study

REYVOW (lasmiditan)
Eli LIlly
INDICATION: Acute treatment of migraine with or without aura in adults.
ADDRESSING UNMET NEED: New option for the acute treatment of migraine that affects one in seven Americans
MECHANISM OF ACTION: Binds with high affinity to the 5-HT1F receptor; presumably exerts its therapeutic effects ithrough agonist effects at the 5-HT1F receptor
EFFICACY:
- 2 randomized, double-blind, placebo-controlled trials, n=3,177 adult patients with history of migraine with and without aura, REYVOW vs, placebo
- Primary Endpoint: % patients whose pain resolved and whose most bothersome migraine symptom (nausea, light sensitivity, or sound sensitivity) resolved two hours after treatment; significantly greater among patients receiving Reyvow.
SAFETY:
- Risk of driving impairment
- Most common side effects: Dizziness, fatigue, a burning or prickling sensation in the skin (paresthesia), sedation
REG PATHWAY: NDA
- Controlled Substance scheduling by DEA pending
- Required pediatric assessments
- Postmarketing requirement: Maternal, fetal, and infant outcomes during pregnancy exposure
SCENESSE (afamelanotide) implant
Clinuvel
INDICATION: To increase pain free light exposure in adult patients with a history of phototoxic reactions from erythropoietic protoporphyria (EPP)
ADDRESSING UNMET NEED: First treatment to increase pain-free light exposure in patients with rare disorder phototoxic reactions from EPP
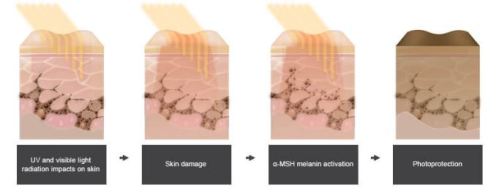
MECHANISM OF ACTION: Synthetic tridecapeptide and a structural analog of α-melanocyte stimulating hormone – melanocortin receptor agonist and binds predominantly to melanocortin 1 receptor.
EFFICACY:
- 2 parallel group clinical trials (n-93, 74), patients with erythropoietic protoporphyria, SCENESSE vs placebo
- First trial primary endpoint: Total number of hours over 180 days spent in direct sunlight between 10 a.m. and 6 p.m. on days with no pain; . 64 hrs vs. 41 hrs
- Second trial primary endpoint: Total number of hours over 270 days spent outdoors between 10 am and 3 pm on days with no pain for which “most of the day” was spent in direct sunlight; 6 hrs vs. 0.75 hrs
SAFETY:
- Most common side effects: implant site reaction, nausea, oropharyngeal pain, cough, fatigue, skin hyperpigmentation, dizziness, melanocytic nevus , respiratory tract infection, somnolence, non-acute porphyria, skin irritation
REG PATHWAY: NDA
- Priority Review, Orphan Drug designation
- Postmarketing requirements: Thorough QT clinical study, Registry based observational exposure cohort study on long-term safety,
Image credits: OraSure, Vertex, Eli Lilly, Clinuvel