
MONARCH external Trigeminal Nerve Stimulation (eTNS) System
NeuroSigma
INDICATION FOR USE: Treatment of pediatric Attention Deficit Hyperactivity Disorder (ADHD) as a monotherapy in patients ages 7 through 12 years old who are not currently taking prescription ADHD medications. The device is used for patient treatment by prescription only and is intended to be used in the home under the supervision of a caregiver during periods of sleep.
ADDRESSING UNMET NEED: Non-drug option for treatment of ADHD in pediatric patients through the use of mild nerve stimulation, a first of its kind
GENERIC DEVICE TYPE:Transcutaneous electrical nerve stimulator for Attention Deficit Hyperactivity Disorder
- Prescription device that stimulates transcutaneously or percutaneously through electrodes placed on the forehead
DEVICE DESCRIPTION:
- Cell-phone sized device generates low-level electrical pulse and connects via wire to a small patch that adheres to a patient’s forehead
- Delivers low-level electrical stimulation to branches of trigeminal nerve, which sends therapeutic signals to the parts of the brain involved in ADHD
- Neuroimaging studies show that eTNS increases activity in brain regions that are known to be important in regulating attention, emotion and behavior
EFFECTIVENESS & SAFETY:
- Clinical trial, n=62 children with moderate to severe ADHD, eTNS vs. placebo
- Primary endpoint: Improvement on clinician-administered ADHD Rating Scale, ADHD-RS
- Subjects using eTNS device had statistically significant improvement in ADHD symptoms vs. placebo group
- At end of week four, the average ADHD-RS score in eTNS decreased from 34.1 to 23.4 points, versusdecrease from 33.7 to 27.5 points in placebo
- Most common side effects: Drowsiness, an increase in appetite, trouble sleeping, teeth clenching, headache and fatigue; no serious adverse events
IDENTIFIED RISKS: Adverse tissue reaction, Injury or discomfort from electrical stimulation, including burns and nerve damage, Misuse that may result in device failure, user discomfort, or injury, Skin irritation or infection from use on broken skin
REGULATORY PATHWAY: De Novo request
- Regulation:21 CFR 882.5898
- Regulation Name: Transcutaneous electrical nerve stimulator for Attention Deficit Hyperactivity Disorder
- Regulatory Class: Class II
- Product Code: QGL
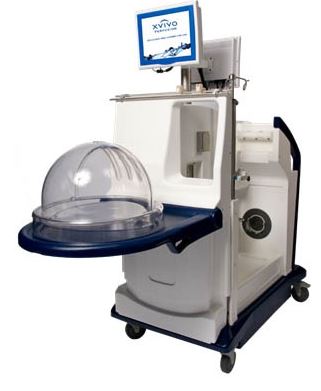
XVIVO PERFUSION SYSTEM
Xvivo Perfusion Inc.
INDICATION FOR USE: For use in flushing and temporary continuous normothermic machine perfusion of initially unacceptable excised donor lungs during which time the ex-vivo function of the lungs can be reassessed for transplantation.
ADDRESSING UNMET NEED:
- Lung transplantation is life-saving treatment for end-stage lung disease
- Many patients die while waiting for suitable lungs for transplant
- Only 15 percent of lungs from deceased donors suitable for transplantation
- New device to temporarily ventilate, oxygenate, and perfuse preservation solution through lungs that were initially thought to be unacceptable for transplant
DESCRIPTION:
- Consists of XPS Perfusion Cart Hardware, fluid path and non-fluid path disposables, XPS Cart Software, and STEEN Solution
- STEEN Solution™ is clear, sterile, non-pyrogenic, non-toxic physiological salt solution containing human serum albumin (HSA) and dextran 40
- Extracellular (low potassium) electrolyte solution with physiological colloid-osmotic pressure (COP) designed for use as a temporary continuous normothermic machine perfusion solution for ex vivo assessment of isolated lungs after removal from the donor
EFFECTIVENESS & SAFETY:
- Study involving 332 sets of donor lungs allocated into three groups
- Control group: Lungs initially deemed suitable for transplant that were provided to 116 recipients after standard preservation
- Second group: Lungs initially deemed unsuitable for transplant and, after being perfused with the Xvivo Perfusion System, were implanted into 110 recipients
- Third group: Perfused with the Xvivo Perfusion System and were still deemed not suitable after the perfusion, and therefore were not implanted into patients
- One-year survival rate: 94% (Control) vs 86.4% (Second and third groups)
- Difference in rates was not deemed to be clinically significant
- Most common adverse events: Acute rejection, bronchial complications, respiratory failure and infections
REGULATORY PATHWAY: PMA
- Classification: III
- Procode: PHO
- Originally granted marketing authorization in 2014 under humanitarian device exemption (HDE), limiting use to maximum of 8,000 patients per year
- PMA pathway allows increased number of lungs to be available for transplant
- Post-Approval requirements: Long-Term Post Approval Studies

COMANECI Embolization Assist Device
Rapid Medical
INDICATION FOR USE: For use in the neurovasculature as a temporary endovascular device used to assist in the coil embolization of wide-necked intracranial aneurysms with a neck width ≤ 10 mm. A wide-necked intracranial aneurysm defines the neck width as ≥ 4 mm or a dome-to-neck ratio < 2
GENERIC TYPE OF DEVICE: Temporary coil embolization assist device
- Prescription device intended for temporary use in the neurovasculature to mechanically assist in the embolization of intracranial aneurysms with embolic coils. The device is delivered into the neurovasculature with an endovascular approach. This device is not intended to be permanently implanted and is removed from the body when the procedure is completed.
RISKS AND MITIGATIONS:
- Infection: Sterilization validation, Pyrogenicity testing, Shelf life testing, Labeling
- Adverse tissue reaction: Biocompatibility evaluation
- Tissue or vessel damage: Non-clinical performance testing, Clinical performance testing, Labeling
- Thromboembolic event: Non-clinical performance testing, Clinical performance testing, Labeling
- Coils ensnarement: Non-clinical performance testing, Clinical performance testing, Labeling
REGULATORY PATHWAY: De Novo request
- Regulation Number: 21 CFR 882.5955
- Regulation Name: Temporary coil embolization assist device
- Regulatory Class: Class II
- Product Code: PUU

NERIVIO MIGRA
Theranica Bioelectronics ltd
INDICATION FOR USE: For acute treatment of migraine with or without aura in patients 18 years of age or older who do not have chronic migraine. It is a prescription use, self-administered device for use in the home environment at the onset of migraine headache or aura.
GENERIC DEVICE TYPE: Trunk and limb electrical stimulator to treat headache
- Device intended to treat headache through the application of electrical stimulation anywhere on the body of the patient apart from the patient’s head or neck through electrodes placed on the skin. The stimulation may be provided transcutaneously or percutaneously
RISKS AND MITIGATIONS:
- Adverse tissue reaction: Biocompatibility evaluation
- Electrical, mechanical, or thermal hazards that may result in user discomfort or injury: Non-clinical performance testing, Electrical, mechanical, and thermal safety testing, Electromagnetic compatibility (EMC) testing, Software verification, validation, and hazard analysis, Labeling
- Interference with other devices: Electromagnetic compatibility (EMC) testing, Labeling
- Software malfunction leading to injury or discomfort: Software verification, validation, and hazard analysis
- Hardware malfunction leading to injury or discomfort: Non-clinical performance testing, Shelf life testing, Labeling
- Use error that may result in user discomfort, injury, or delay treatment for headaches: Labeling
REGULATORY PATHWAY: De Novo request
- Regulation Number: 21 CFR 882.5899
- Regulation Name: Trunk and limb electrical stimulator to treat headache
- Regulatory Class: Class II
- Product Code: QGT

ZIKV Detect 2.0 IgM Capture ELISA
InBios
INDICATION FOR USE: Intended for the qualitative detection of Zika virus IgM
antibodies in human sera for the presumptive clinical laboratory diagnosis of Zika virus infection.
The assay is intended for use only in patients with clinical signs and symptoms consistent with Zika virus infection, and/or CDC Zika virus epidemiological criteria (e.g., history of residence in or travel to a geographic region with active Zika transmission at the time of travel, or other epidemiological criteria for which Zika virus testing may be indicated). Assay results are for the presumptive detection of IgM antibodies to Zika virus (ZIKV). Positive results must be confirmed by following the latest CDC guidelines for the diagnosis of Zika virus infection.
ADDRESSING UNMET NEED:
- 1st Commercial serology kit to receive FDA Marketing Authorization
- Previous tests for detecting Zika virus IgM antibodies—including the ZIKV Detect 2.0 IgM Capture ELISA—had been authorized only for emergency use under the FDA’s Emergency Use Authorization (EUA) authority
GENERIC DEVICE TYPE: Zika virus serological reagents
- In vitro diagnostic devices that consist of antigens or antibodies for the detection of Zika virus or Zika antibodies in human specimens from individuals who have signs
and symptoms consistent with Zika virus infection and/or epidemiological risk factors. The detection aids in the diagnosis of current or recent Zika virus infection or serological status. Negative results obtained with this test do not preclude the possibility of Zika virus infection, past or present. Positive results should be interpreted with consideration of other clinical information and laboratory findings and should not be used as the sole basis for treatment or other patient management decisions.
RISKS AND MITIGATIONS:
- Risk of false results: Device description, performance characteristics, validation procedures, labeling
- Failure to correctly interpret test results: Device description, performance characteristics, Labeling
- Failure to correctly operate the device: Device description, performance characteristics, validation procedures, Labeling
REGULATORY PATHWAY: De Novo request (previously authorized via EUA)
- Regulation Number: 21 CFR 866.3935
- Regulation Name: Zika Virus Serological Reagents
- Regulatory Class: Class II
- Product Code: QFO
Image credits: NeuroSigma, Xvivo, Rapid Medical, Theranica, InBios