 Technical Considerations for Additive Manufactured Medical Devices
Technical Considerations for Additive Manufactured Medical Devices
Outline technical considerations with Additive Manufacturing (3D printing) and recommendations for testing and characterization
- Builds object by sequentially building 2D layers, joining each to the layer below to rapidly produce alternative designs
- Process Flow

Design and Manufacturing Process Considerations
- Overall Device Design
- Patient-Matched Device Design
- Software Workflow
- Material Control
- Post-Processing
- Process Validation and Acceptance
- Quality Data
Device Testing Considerations
- Device Description
- Mechanical Testing
- Dimensional Measurements
- Material Characterization
- Removing Manufacturing Material Residues and Sterilization
- Biocompatibility
Labeling
- patient identifier, use, final design iteration or version used to produce the device
 FDA Categorization of Investigational Device Exemption (IDE) Devices to Assist the Centers for Medicare and Medicaid Services (CMS) with Coverage Decisions
FDA Categorization of Investigational Device Exemption (IDE) Devices to Assist the Centers for Medicare and Medicaid Services (CMS) with Coverage Decisions
Efficient CDRH categorization of investigational medical devices to support CMS’s Medicare coverage (reimbursement) determinations
- Process and information to determine appropriate IDE category
- Change of assigned category
FDA Interpretation of Medicare Coverage Categories A and B
- Category A-Experimental: No PMA approval, 510(k) clearance, or De Novo, being studied for new indication/new intended use; prior information does not resolve initial safety/effectiveness questions
- Category B- Nonexperimental/Investigational: No PMA approval, 510(k) clearance, or De Novo, being studied for a new indication/new intended use; prior information does resolve initial safety/effectiveness questions
Considerations When Changing from Category A to B – data to support
- Peer-reviewed studies on similar device
- Premarket or postmarket data from ex-US studies with similar device
- Commercialization of similar device
- Preliminary clinical data
- Additional non-clinical data

 Clinical and Patient Decision Support Software
Clinical and Patient Decision Support Software
FDA’s regulatory oversight of:
(1) clinical decision support (CDS) software intended for healthcare professionals
(2) patient decision support (PDS) software intended for patients and caregivers who
are not healthcare professionals.
Scope of FDA oversight of CDS/PDS software:
- do not meet the definition of device as amended by the Cures Act
- may meet definition of device but will not require premarket clearance and premarket approval
- will require regulatory oversight
FDA Definitions:
CDS: Software functions that are considered as device:
- intended to acquire, process, analyze a medical image/signal from in
vitro diagnostic device or pattern or signal from signal acquisition system - intended for displaying, analyzing, or printing medical information
- intended for supporting or providing recommendations to health care
professional
Function excluded from the definition of device with additional criterion
- intended for enabling health care professional to independently review basis for software recommendations and does not rely primarily on recommendation
for clinical diagnosis or treatment decision
Examples of functions that are and are not considered as devices provided
PDS: Low risk devices and fall outside functionalities of regulatory oversight
- enforcement discretion policy
- software function should clearly explain:
- purpose or intended use
- intended user (e.g., patient, non-health care professional caregiver)
- inputs used to generate recommendation
- rationale or support for recommendation
Examples provided

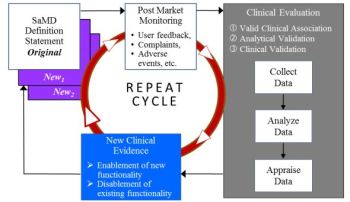
 Software as a Medical Device (SAMD): Clinical Evaluation
Software as a Medical Device (SAMD): Clinical Evaluation
Global regulatory framework and principles for SaMD
- adopts internationally converged principles agreed upon by IMDRF
- FDA adoption of principles
Clinical Evaluation of SaMD
- Valid Clinical Association
- Analytical / Technical Validation
- Clinical Validation of a SaMD

General Principles and Context of Clinical Evaluation Process
- Definition Statement and Category
- Clinical Evaluation Processes
Clinical Evaluation Process Flow Chart
- Considerations for Generating and Assessing Evidence
Importance of Independent Review of Clinical Evaluation
Continuous Learning Leveraging Real World Performance Data

Changes to Existing Medical Software Policies Resulting from Section 3060 of the 21st Century Cures Act
Section 3060(a) of 21st Century Cures Act amended section 520 of (FD&C Act) removing certain software functions from definition of devic
- affects FDA’s guidances related to medical device software.
Level 2 updates to be made to following guidance documents:
- General Wellness: Policy for Low Risk Devices
- Mobile Medical Applications
- Off-The-Shelf Software Use in Medical Devices
- Medical Device Data Systems, Medical Image Storage Devices, and Medical Image
Communications Devices
Withdrawing following guidance document:
- Submission of Premarket Notifications for Medical Image Management Devices
Interpretation of Cures Act and modifications to existing guidances:
- Software Function Intended for Administrative Support of Health Care Facility
- Software Function Intended for Maintaining or Encouraging Healthy Lifestyle
- Software Function Intended to Serve as Electronic Patient Records
- Software Function Intended for Transferring, Storing, Converting Formats, Displaying Data and Results.