FDA Approvals: GAZYVA and AFINITOR
FDA Brief: Week of Feb 22, 2016

GAZYVA (obinutuzumab) Injection
Genentech, Inc, San Francisco, CA, USA
Indication: In combination with bendamustine followed by GAZYVA monotherapy, for the treatment of patients with follicular lymphoma (FL) who relapsed after, or are refractory to, a rituximab-containing regimen
Reg. Pathway: Supplemental BLA, (previously approved for chronic lymphocytic leukemia), Priority Review
Efficacy:
- Single open-label, multicenter, randomized study (n=321); patients with FL who had no response to or have progressed during or within 6 months of rituximab or a rituximab-containing regimen; GAZYVA plus bendamustine vs. bendamustine alone
- 6 cycles of Gazyva plus bendamustine followed by continued Gazyva monotherapy for up to 2 years with 6 cycles of bendamustine therapy
- Primary Endpoint: Progression Free Survival (PFS) by independent review. Median PFS 13.8 months for bendamustine alone vs . not reached in the Gazyva plus bendamustine, p-value < 0.0001
- Median Overall Survival not yet reached in either arm


Safety:
- Serious adverse reactions: Febrile neutropenia, neutropenia, infusion related reactions, sepsis, pneumonia and pyrexia
- Most common adverse reactions: Infusion reactions, neutropenia, nausea, fatigue, cough, diarrhea, constipation, pyrexia, thrombocytopenia, vomiting, upper respiratory tract infection, decreased appetite, arthralgia, sinusitis, anemia, asthenia and urinary tract infection.

AFINITOR ( everolimus) Tablet
Novartis, East Hanover, NJ, USA
Indication: Treatment of adult patients with progressive, well-differentiated, non-functional neuroendocrine tumors (NET) of gastrointestinal (GI) or lung origin with unresectable, locally advanced or metastatic disease
Reg Pathway: Supplemental NDA (previously approved for breast cancer and several other cancer indications)
Efficacy:
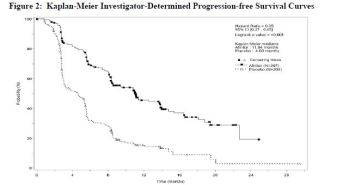
- Single, multicenter, randomized (2:1), n=302, patients with unresectable, locally advanced or metastatic, well differentiated (low or intermediate grade), non-functional (no current or prior history of carcinoid symptoms), neuroendocrine tumors (NET) of gastrointestinal or lung origin
- Afinitor plus best supportive care (BSC) vs. placebo plus BSC
- Primary Endpoint : Improvement in progression-free survival (PFS)
- Median PFS : 11 mo. vs. 3.9 mo., p <0.001
- Overall Response Rates: 2% vs. 1%
- Overall Survival: No statistically significant difference
Safety:
- Serious adverse reactions: In 42% of Afinitor-treated patients and included 3 fatal events (cardiac failure, respiratory failure, and septic shock
- Most common adverse reactions: Stomatitis, infections, diarrhea, peripheral edema, fatigue and rash
- Most common laboratory abnormalities: anemia, hypercholesterolemia, lymphopenia, elevated aspartate transaminase and fasting hyperglycemia